O blog da AWS
Diagnóstico de leucemia por machine learning no Laboratório de Leucemia de Munique com o Amazon SageMaker
- Manipulação dos arquivos de chamada da variante de NGS para gerar recursos úteis para classificação
- Exame da separabilidade das diferentes classes, com base em métricas de similaridade
- Constatação do melhor modelo para a classificação e interpretabilidade do subtipo de leucemia
- Conclusões da colaboração
Introdução
A leucemia consiste em um conjunto de cânceres no sangue que apresentam um risco significativo à saúde, causando mais de 310 mil mortes anualmente em todo o mundo, de acordo com o estudo da Global Burden of Disease de 2018. O diagnóstico e o tratamento são altamente complicados devido à heterogeneidade; há 31 entidades de leucemia (subtipos) definidas pela Organização Mundial da Saúde desde 2017.
Atualmente, a leucemia é diagnosticada usando uma combinação de métodos. Esses métodos requerem equipamentos complexos, além de cientistas e técnicos de laboratório clínico altamente qualificados (recursos escassos), o que aumenta o tempo de resposta, bem como os custos. Em média, desde o recebimento da amostra até o relatório, pode levar até dez dias.
Uma mudança de paradigmas na metodologia do diagnóstico
O sequenciamento de nova geração (NGS), uma tecnologia de sequenciamento massivo paralelo de DNA, capaz de ler um genoma humano inteiro em um dia, tem se mostrado uma grande promessa para identificar subtipos de leucemia, bem como prescrever um tratamento mais direcionado e eficaz.
Prof. Dr. Torsten Haferlach, CEO do MLL, afirmou: “Para estabelecer essa mudança de paradigma, de uma revisão prioritariamente manual de características morfológicas feita por especialistas experientes a uma abordagem algorítmica objetiva usando recursos baseados em genética molecular, o MLL fez uma parceria com o Laboratório de Soluções da Amazon Machine Learning para lidar com esse feito extraordinário. Esse trabalho aumentará os casos de cura e prolongará o tempo de vida de todos os pacientes”.
Classificação do câncer com base em dados de NGS usando machine learning
O Laboratório de Soluções do Amazon Machine Learning aceitou o desafio de auxiliar o MLL no desenvolvimento de um modelo inovador de suporte à decisão clínica, baseado em um conjunto de dados de sequenciamento de nova geração (NGS). Os dados de NGS são altamente dimensionais e geralmente são compostos de quantidades muito pequenas de dados de treinamento de qualidade. O MLL, tendo concluído o projeto dos 5 mil genomas, tem um dos maiores conjuntos de dados nesse domínio. A maioria das amostras dos 5 mil pacientes envolvidos, portadores de leucemia, foi sequenciada pelo sequenciamento completo do genoma (WGS) e sequenciamento completo do transcriptoma (WTS), e diagnosticada usando os métodos ortogonais de diagnóstico padrão-ouro da OMS.
Preparação dos dados
Com o escopo global da pesquisa do MLL, é imperativo que os dados sejam mantidos em serviços de nuvem que sejam seguros e estejam em conformidade com os regulamentos de privacidade. A AWS está disponível em muitos países e regiões, o que proporciona praticidade e economia aos clientes. Todos os dados de clientes podem ser armazenados e acessados em qualquer área da AWS. Nesse caso específico, os dados do MLL foram armazenados em buckets do Amazon S3, na região selecionada pelo cliente, em Frankfurt, para atender às requisições de dados de assistência médica.
Os dados de NGS do DNA (sequenciamento completo do genoma) e RNA (sequenciamento completo do transcriptoma) foram inicialmente processados pelo MLL usando um pipeline personalizado. Do WGS, vários tipos de variantes podem ser extraídas: variante de nucleotídeo único (SNV), variantes estruturais (SV) e variantes de número de cópia (CNV). Os dados de SV e CNV ficam em arquivos VCF, que têm uma estrutura complexa. Arquivos VCF incluem informações de anotação funcional da variante (variant call), como o cromossomo e a localização do segmento da variante, a área de leitura e os critérios de filtragem da variante. Arquivos VCF de dados de SV e CNV não podem ser facilmente importados para formatos que possam ser ingeridos por modelos de machine learning. É necessário um processamento adicional para extrair e agregar características com base no nível de granularidade escolhido, que pode estar no nível de genes, bandas cromossômicas ou cromossomos.
Do pipeline do WTS, dados de expressão gênica (GE) e fusão gênica (GF) podem ser obtidos. A vantagem dos dados de GE é que já possuem um formato tabular onde cada linha é um paciente e cada coluna é o valor de expressão de um gene, portanto, não há mais transformações, mas apenas um procedimento rápido de normalização que deve ser realizado. A GF é mais parecida com os arquivos obtidos para o WGS, então transformações semelhantes são realizadas.
Estabelecemos um pipeline que pega os arquivos de dados de cada paciente e os transforma em vetores de recursos para cada modalidade. Para arquivos SV, extraímos cinco tipos de variantes: inserções, deleções, duplicações, translocações e inversões no nível do gene para segmentos curtos. Para segmentos mais longos, extraímos recursos com base no nível da banda ou do cromossomo. Essa abordagem também foi usada para arquivos CNV. Para dados do WTS, a expressão gênica é extraída na forma de contagens de leitura e é normalizada seguindo o método de normalização de média aparada ponderada de valores M (TMM). Como resultado, geramos mais de 70 mil elementos das cinco tabelas originais (SNV, SV, CNV, expressão gênica, fusão gênica). Os dados ausentes foram imputados de forma diferente, dependendo do tipo de origem. Para alguns tipos, usamos o valor mais baixo (CNV) ou zero (SNV, SV, expressão gênica). No final do processamento de dados, cada paciente possui o mesmo número de elementos, que podem ser usados como entrada para um modelo de machine learning.
Avaliamos várias estratégias para combinar essas diferentes modalidades, inclusive modelos preparados de empilhamento ou agrupamento em cada modalidade individual. Entretanto, o manuseio separado desses tipos de dados não leva em consideração a observação de que existem interações não lineares entre características de diferentes modalidades (por exemplo, conexões entre amplificação/deleção do número de cópias e regulação ascendente ou descendente do nível de expressão gênica, SVs associados à expressão gênica aberrante). O modelo deve levar em conta essas interações para ter um bom desempenho, e é por isso que a associação de todos os tipos de elementos para permitir que o modelo aprenda as interações entre eles levou à maior precisão preditiva.

Figura 1: visão geral dos arquivos processados para obtenção de um conjunto tabular de dados para treinamento. “||” refere-se à associação dos vetores obtidos para cada tipo de arquivo.
Análise de pré-modelo
O conjunto de dados do MLL contém pacientes com 30 subtipos diferentes de leucemia. Alguns dos subtipos são muito mais frequentes do que outros, resultando em um conjunto de dados altamente desequilibrado.
 Figura 2: número de pacientes por subtipo de leucemia.
Figura 2: número de pacientes por subtipo de leucemia.
Com todos os diferentes tipos de dados convertidos para o formato tabular, iniciamos um processo de engenharia de recursos que envolveu a colaboração com especialistas do MLL. Eles pesquisaram a literatura para encontrar biomarcadores importantes associados a cada subtipo de leucemia. Com isso em mente, projetamos um processo para agregar e combinar os recursos originais em biomarcadores.
O conjunto de dados final tinha cerca de 4.500 linhas representando pacientes e 800 colunas contendo os dados de biomarcadores extraídos, combinados com outros elementos importantes para cada tipo de dado (CNV, SNV, SV etc.) fora do conjunto de recursos do biomarcador original. Em outras palavras, o genoma e o transcriptoma de cada paciente foram representados por um vetor com 800 entradas.
Entre os 30 subtipos de leucemia, alguns deles estão mais próximos uns dos outros do que dos demais. Por exemplo, MPAL, LMA e BCP-ALL são muito semelhantes; às vezes, o perfil genético de um paciente com MPAL pode ser mais semelhante ao de um paciente com BCP-ALL do que ao de outro paciente com MPAL. Isso representa um desafio, tanto para o algoritmo de machine learning quanto para especialistas humanos, no que diz respeito à sua diferenciação. Para investigar a semelhança entre os subtipos de leucemia, calculamos as distâncias entre cada paciente e o restante das amostras, e registramos quais eram os dois semelhantes mais próximos. Em seguida, agregamos os semelhantes por subtipo de leucemia e descobrimos que, de fato, em algumas classes, menos de 20% dos pacientes desse subtipo são muito semelhantes entre si. Como mostra a figura 3: Se encontrarmos o círculo MPAL, poderemos ver que apenas 13% de todos os pacientes com MPAL são semelhantes entre si e que a maioria dos pacientes semelhantes pertence a LMA e BCP-ALL.
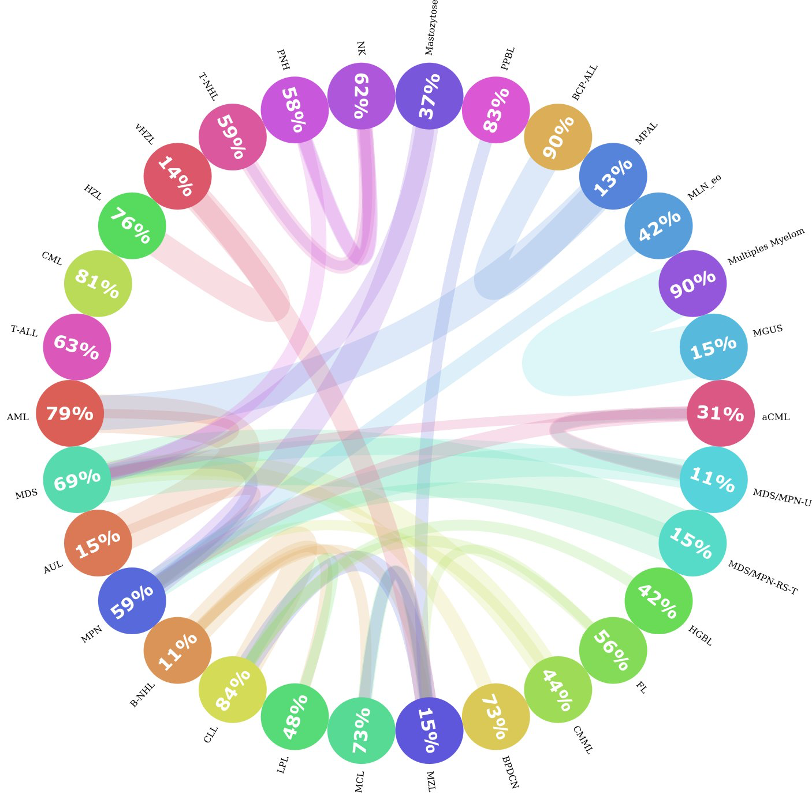
Figura 3: gráfico circos de similaridade de subtipos. Esta figura mostra como os subtipos de leucemia são semelhantes entre si. Cada círculo representa um subtipo de leucemia e o número dentro do círculo captura a porcentagem de vizinhos do mesmo subtipo. Se houver uma parcela significativa (>= 10%) de pacientes que são mais semelhantes aos pacientes de outro subtipo (entidade), então uma conexão será traçada (linha colorida) de um organismo para o outro. A espessura da linha representa o tamanho da parcela de pacientes semelhante a outros grupos. A cor da linha representa a direção da conexão, ou seja, o paciente de um organismo pertencente a outros grupos; é representado com a cor da entidade original. Por exemplo, os pacientes da classe MGUS eram mais semelhantes aos pacientes com mieloma múltiplo. Mas não há conexões significativas no sentido inverso.
Modelagem
Para prever subtipos de leucemia de pacientes, treinamos um classificador multiclasse. Com o classificador e o canal de extração de elementos, criamos um protótipo de sistema que determina automaticamente o subtipo de leucemia do paciente com base em dados do WGS e WTS, usando o Amazon SageMaker Notebooks. O Amazon SageMaker é uma plataforma de machine learning em nuvem que pode ser usada para criar, treinar e implantar modelos para praticamente qualquer caso. As instâncias do Notebook fornecem ambientes flexíveis para criar modelos de machine learning, economizando tempo e esforço dos usuários com o gerenciamento da infraestrutura computacional subjacente.
Usamos o LightGBM, um algoritmo de aumento de gradiente, para desenvolver nosso primeiro classificador. Também usamos trabalhos de Otimização de Hiperparâmetros (HPO) do SageMaker para ajustar os hiperparâmetros do nosso modelo, uma abordagem que identifica automaticamente a configuração ideal de qualquer algoritmo sem esforço manual, utilizando a otimização bayesiana. Usando o HPO, o desempenho do nosso modelo LGBM atingiu uma precisão de 82% na validação cruzada de 5 dobras. Para alguns organismos com biomarcadores exclusivos, a precisão é muito alta. A CML tem um gene de fusão BCR-ABL quase exclusivo como biomarcador e o modelo LGBM atinge 97% de precisão para a LMC.
Porém, para organismos com bem poucos pacientes, com forte semelhança com outros organismos ou que requerem uma análise laboratorial extra durante o diagnóstico tradicional, o modelo não é tão eficaz. Para lidar com amostras pequenas e desequilibradas, você pode usar a geração de dados sintéticos. Tentamos métodos diferentes, inclusive LORAS e ADASYN, mas não vimos nenhuma melhoria no modelo.
Modelos que utilizam aprendizado simples também foram usados em problemas de classificação com bem poucas amostras por categoria. Implementamos uma arquitetura de aprendizado simples para a previsão do tipo de doença, mas não vimos nenhuma melhoria. Isso provavelmente se deve ao desafio de entender a distribuição de um tipo de leucemia com o número muito pequeno de pacientes que tínhamos disponíveis para cada tipo.
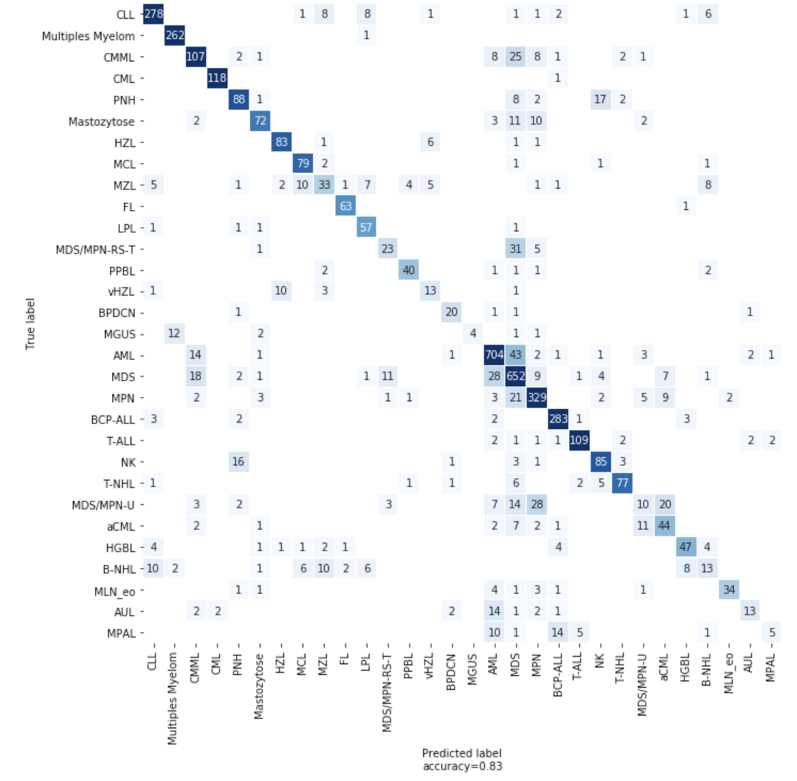
Figura 4: matriz de confusão mostrando o número de previsões corretas e incorretas do modelo
Explicação
Quando um modelo de machine learning é usado para fazer uma previsão, entender por que a previsão foi feita é tão importante para os pesquisadores do MLL quanto a própria previsão. Para entender por que o modelo está fazendo previsões específicas, fizemos uso da biblioteca python SHAP. Com essa biblioteca, podemos analisar uma previsão do modelo treinado e obter informações sobre os recursos usados para fazer a classificação. Aplicamos o SHAP ao nosso modelo LGBM e analisamos os impactos dos recursos tanto no nível da amostra quanto no nível global. A Figura 5 ilustra uma aplicação do SHAP no nível do paciente para dois indivíduos diagnosticados com LMC em comparação com a coorte corretamente prevista da LMC. Usando um gráfico de decisão, podemos observar quais recursos são os contribuintes mais importantes para a previsão do modelo.

Figura 5: gráfico de força SHAP para um paciente com LMC previsto corretamente, e um gráfico de decisão para cada paciente com LMC, 117 previstos corretamente (linhas amarelas) e um previsto incorretamente (linha vermelha). As características são mostradas no eixo y, em ordem de importância média para todos os pacientes com LMC; observe os elementos conhecidos de BCR/ABL1 na parte superior. Esses recursos empurraram o classificador para a LMC em ambos os pacientes, mas para o paciente que foi previsto incorretamente, as características de expressão gênica fizeram com que o classificador previsse BCP-ALL. Esses gráficos de decisão são úteis para entender o quanto cada elemento contribui para a classificação; observe que o eixo x é “log-odds”, portanto, um elemento que gera uma saída de 0 a 10 é extremamente mais importante do que um que gera -5 a 0.
Conclusões e panorama
Colaboração com especialistas no domínio: isso tem sido crucial e continuará sendo uma parte fundamental na aplicação de machine learning e IA no domínio médico. Além de compartilhar o maior conjunto de dados, com a mais alta qualidade do seu tipo, o MLL facilitou a criação do modelo de machine learning, contribuindo com sua experiência significativa de domínio a cada passo ao longo do caminho. No início do projeto, codificamos nosso espaço de elementos no nível do gene e só alcançamos uma precisão de 67% em 30 previsões de subtipo de leucemia. Ao usar biomarcadores e características genéticas selecionados manualmente do MLL, alimentando o modelo com o que já é conhecido sobre esse domínio por especialistas humanos, alcançamos 77% de precisão. Posteriormente, adicionamos mais características ao espaço de características e aumentamos ainda mais o desempenho do modelo em 5%.
Usando as ferramentas certas: trabalhar com o SageMaker acelerou significativamente esse projeto. Como o SageMaker é uma plataforma totalmente gerenciada e integrada para ML, ele permitiu que a equipe se concentrasse no exercício da ciência de dados sem ter que se preocupar com o gerenciamento da infraestrutura. O recurso integrado de ajuste automático aumentou a precisão do modelo sem ter que ajustar manualmente a sua arquitetura ou os hiperparâmetros, economizando tempo e otimizando o modelo.
Interpretabilidade e capacidade de explicação: o MLL continua inovando e melhorando sua abordagem, além de trabalhar sua visão sobre o diagnóstico ultrarrápido de leucemia, que é altamente preciso e algoritmicamente objetivo, beneficiando pacientes em todo o mundo. Nas palavras de Benjamin Bell, citadas pelo Prof. Haferlach durante uma palestra: “…a IA não substituirá os médicos, mas os aprimorará, permitindo que pratiquem uma medicina melhor, mais precisa e eficiente”.
Conclusão
A missão do MLL é melhorar o atendimento de pacientes com leucemia e linfoma por meio de diagnósticos de ponta. Para fazer isso, o laboratório usa uma abordagem interdisciplinar combinando seis disciplinas (citomorfologia, imunofenotipagem, análise cromossômica, hibridização in situ fluorescente (FISH), genética molecular e bioinformática), resultando em um relatório abrangente de laboratório integrado.
“Em geral, podemos distinguir subtipos de doenças por meio de métodos fenotípicos, como citomorfologia, imunofenotipagem e técnicas genéticas. No futuro, as técnicas moleculares serão os métodos mais importantes para nos ajudar a capturar todas essas informações”, afirmou o Prof. Haferlach. “Hoje, o diagnóstico genético depende da citogenética e da análise de mutações. Em breve, faremos o sequenciamento completo do genoma e do transcriptoma.”
O sequenciamento completo do genoma pode identificar mais variantes estruturais e copiar alterações numéricas do que são avaliáveis por meio da análise cromossômica, e também pode ser usado para detecção e análise de mutações, explicou. “Pode haver um exame que faça tudo isso”, ele estimou. “Se também incluirmos perfis de expressão gênica em nossos diagnósticos, poderemos até detectar características que atualmente são determinadas por imunofenotipagem e citomorfologia. Esta é a nossa visão do futuro.”
“Somos professores e pesquisadores”, o Dr. T. Haferlach apoiou. “Queremos estar sempre na vanguarda do que está por vir.” Esse desejo levou os fundadores do MLL a adotar e incorporar totalmente a tecnologia no seu laboratório e firmar parceria com a AWS em busca de inovação.
Para saber mais sobre genômica na Nuvem AWS, veja thinkwithwp.com/health/genomics.
Este artigo foi traduzido do Blog da AWS em Inglês.